
VAD-MM/GBSA Description
|
MM/GBSA is widely used in end-point binding free energy prediction in structure-based drug design. Here, combining with machine-learning optimization, we present a refinement version of MM/GBSA named Variable Atomic Dielectric MM/GBSA (VAD-MM/GBSA) that exhibits improved accuracy for predicting binding affinities of diverse protein-ligand systems and is promising to be used in post-processing of structure based virtual screening. Simulated annealing-based optimization is used to search the optimal atomic dielectric constant distributions, which are correlative with 87 descriptors of the ligand and the pocket of protein by XGBoost regression. Click here for more information. If you have any questions, please contact us. |
Submitting Jobs
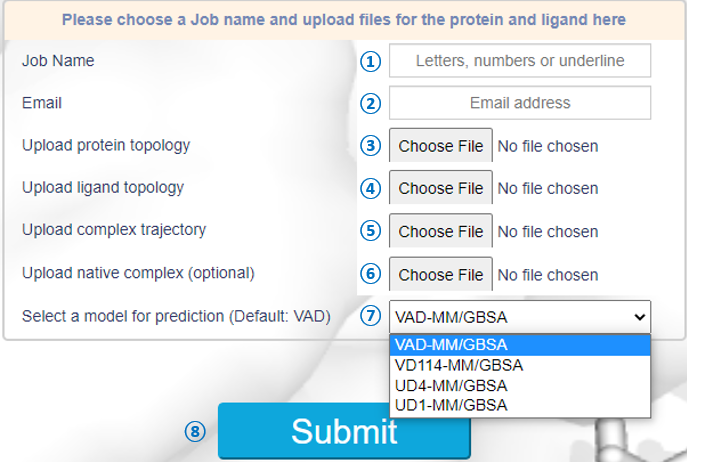
|
① Input your jobname with letters, numbers or underline. ② Input your email address to receive the results. ③ Upload an Amber topology (.prmtop) for the protein. ④ Upload an Amber topology (.prmtop) for the ligand. ⑤ Upload an Amber NetCDF trajectory (.nc) for the protein-ligand complex. 25 (or 50) MD snapshots in the trajectory are supported. ⑥ Upload an Amber NetCDF file (.nc) for the protein-ligand native structure (optional). If not provided, the last frame in the trajectory will be used as the native structure. ⑦ Select a model for prediction (VAD-MM/GBSA default). ⑧ Click here to submit your job. |
| Note: |
|
Job Output
| Once your job is completed, an email with the result calculated by our VAD-MM/GBSA method will be sent to you, and then you can extract the information you are interested from the result. |
|
Queue
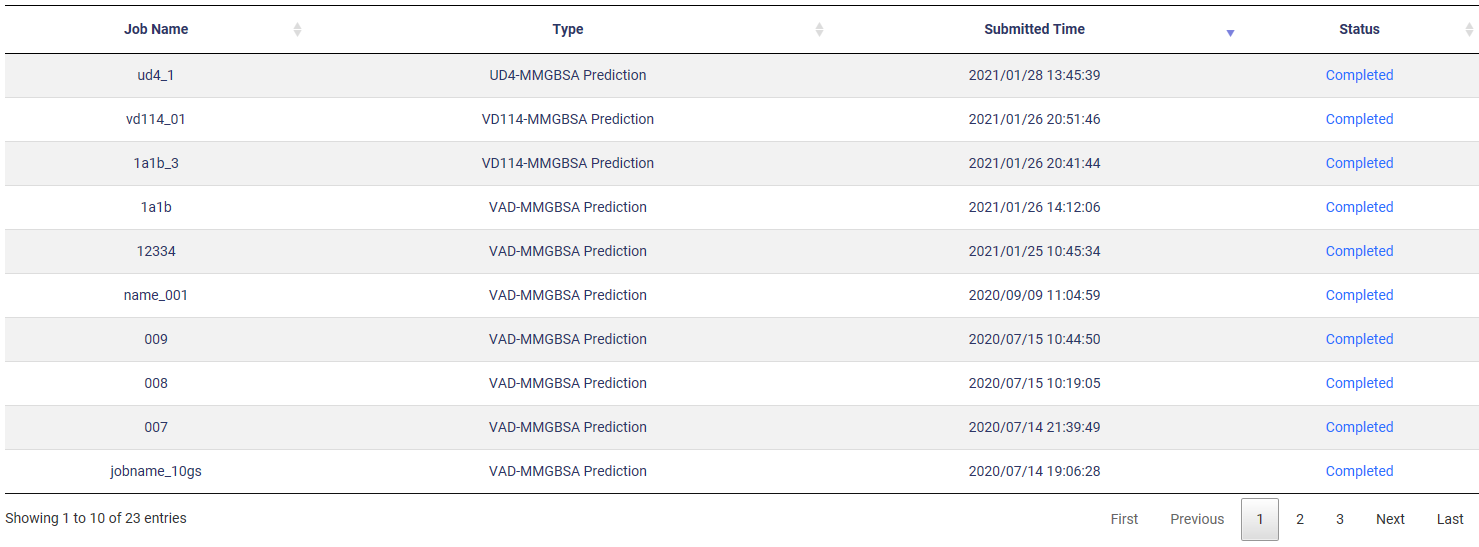
|
| Name: your jobname |
|
Type: function type |
| Submitted Time: the system time when you submitted your job |
Status:
|