1NLJ
Target information
- RCSB PDB
- 1NLJ
- Title
- CRYSTAL STRUCTURE OF THE CYSTEINE PROTEASE HUMAN CATHEPSIN K IN COMPLEX WITH A COVALENT AZEPANONE INHIBITOR
- Method
- X-RAY DIFFRACTION
- Resolution
- 2.4
- Classification
- HYDROLASE
- Organism
- Homo sapiens
- Protein
- Cathepsin K (P43235) Looking for covalent inhibitors of this target ?
- Year
- 2003
- Publication Title
- Azepanone-Based Inhibitors of Human and Rat Cathepsin K
- Abstract
-
The synthesis, in vitro activities, and pharmacokinetics of a series of azepanone-based inhibitors of the cysteine protease cathepsin K (EC 3.4.22.38) are described. These compounds show improved configurational stability of the C-4 diastereomeric center relative to the previously published five- and six-membered ring ketone-based inhibitor series. Studies in this series have led to the identification of 20, a potent, selective inhibitor of human cathepsin K (K(i) = 0.16 nM) as well as 24, a potent inhibitor of both human (K(i) = 0.0048 nM) and rat (K(i,app) = 4.8 nM) cathepsin K. Small-molecule X-ray crystallographic analysis of 20 established the C-4 S stereochemistry as being critical for potent inhibition and that unbound 20 adopted the expected equatorial conformation for the C-4 substituent. Molecular modeling studies predicted the higher energy axial orientation at C-4 of 20 when bound within the active site of cathepsin K, a feature subsequently confirmed by X-ray crystallography. Pharmacokinetic studies in the rat show 20 to be 42% orally bioavailable. Comparison of the transport of the cyclic and acyclic analogues through CaCo-2 cells suggests that oral bioavailability of the acyclic derivatives is limited by a P-glycoprotein-mediated efflux mechanism. It is concluded that the introduction of a conformational constraint has served the dual purpose of increasing inhibitor potency by locking in a bioactive conformation as well as locking out available conformations which may serve as substrates for enzyme systems that limit oral bioavailability.
- External Link
- RCSB PDB
Ligand information
- HET
- 2CA
- Chain ID
- A
- HET Number
- 300
- Molecular Formula
- C26H30N4O6S
- Structure
-
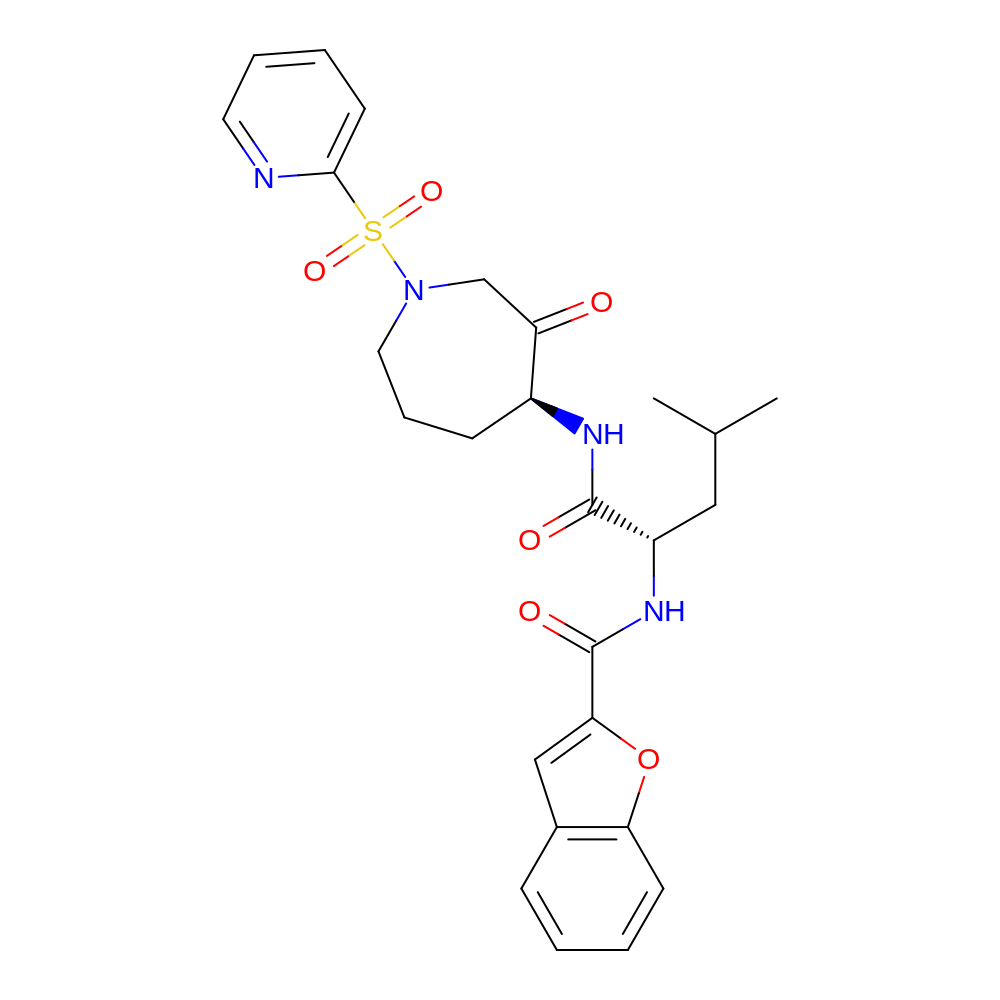
- IUPAC Name
- N-[(1S)-3-methyl-1-[[(4S)-3-oxo-1-(2-pyridylsulfonyl)azepan-4-yl]carbamoyl]butyl]benzofuran-2-carboxamide
- InChI
- InChI=1S/C26H30N4O6S/c1-17(2)14-20(29-26(33)23-15-18-8-3-4-10-22(18)36-23)25(32)28-19-9-7-13-30(16-21(19)31)37(34,35)24-11-5-6-12-27-24/h3-6,8,10-12,15,17,19-20H,7,9,13-14,16H2,1-2H3,(H,28,32)(H,29,33)/t19-,20-/m0/s1
- InChI Key
- VBPPNJCVXGAZDD-PMACEKPBSA-N
- Canonical SMILES
- c1cccc(c12)cc(o2)C(=O)N[C@@H](CC(C)C)C(=O)N[C@H]3CCCN(CC3=O)S(=O)(=O)c4ccccn4
- Bioactivity data
- No bioactivity data available for this ligand.
Covalent Binding
- Warhead
- Carbonyl
- Reaction Mechanism
- Nucleophilic Addition
- Residue
- CYS : 25
- Residue Chain
- A
- Interactions
- Pharmacophore Model