Ready-to-dock benchmark database
Based on the 2P2I database (version 2.0, 28-03-2018),we construct a ready-to-dock benchmark database which contains 184 PPI-inhibitor systems. For each system, the crystal structure of protein-ligand complex was derived from Protein Data Bank (PDB) and then was prepared using the prepwizard module in the Schrödinger suites, during which appropriate bond orders were assigned to all residues, and hydrogen atoms were added using the default parameters. The hydrogen bond network was optimized by enabling the ‘flipping’ of side chains which were ambiguously defined by electron density (Asn, Gln, and His). Then a restrained minimization was performed with a root-mean-squared deviation (RMSD) termination criterion of 0.3 Å on the protein heavy atoms with OPLS 3 force field. Finally, each prepared complex was split into protein, ligand, waters and ions manually in the Maestro.
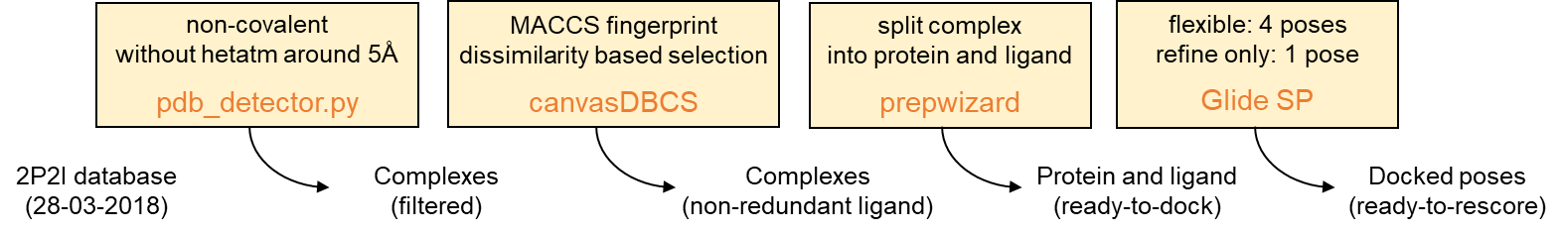 Figure 1. Benchmark database preparation workflow
Figure 1. Benchmark database preparation workflow
Download: ready-to-dock.zip
Ready-to-rescore benchmark database
Based on our ready-to-dock benchmark database, Glide docking program in standard-precision (SP) mode was utilized to generate the binding poses of inhibitors. It is necessary to guarantee that there will be at least one near-native pose among the docking poses for every ligand for the subsequent pose ranking performance benchmark. Thus, both flexible and refine-only sampling algorithms were used to generate up to four and one docking pose, respectively, which means that the maximum number of docking poses of each ligand is five. Finally, 910 docking poses were generated for 168 PPI-inhibitor systems.
Download: ready-to-rescore.zip